The off-diagonal sampling procedure can be simplified for the
application to the electronic excitations in hydrogen. For all
temperatures under consideration, the thermal de Broglie wave length
of the protons is small compared to the inter-particle spacing and also
much smaller than that of the electrons. Therefore, one can make the
protons classical within a first approximation. In this case, one
replaces the proton path by a point particle and needs only one open
electron path. The pairs ![]() and
and ![]() in Eq. 5.15 are then given by
the separation of the ends of the electron paths and the proton. This
has the advantage that one can average over all protons in a many
proton simulation discussed in section 5.4.3.
in Eq. 5.15 are then given by
the separation of the ends of the electron paths and the proton. This
has the advantage that one can average over all protons in a many
proton simulation discussed in section 5.4.3.
|
In the PIMC simulation, one calculates the matrices for a certain
number of angular momentum states using the average given by
Eq. 5.16. We kept the matrices for ![]() and used
a uniform grid in real space with 50 points from
and used
a uniform grid in real space with 50 points from ![]() to
to
![]() . This includes an approximation because one does not
consider the cubic symmetry of the simulation cell. In the limit of a
large cell, this approximation becomes more and more accurate because
the majority of the occupied natural orbitals only extends over a
fraction of the simulation cell and therefore is not affected by the
boundary conditions. The justification for using this basis is that
one is generally interested in systems where the natural orbitals are
determined by the interactions rather than by boundary effects. However, in
systems where those are important, another basis that includes the cubic
symmetry is more appropriate. Suggestions have been made by
Shumway (1999).
. This includes an approximation because one does not
consider the cubic symmetry of the simulation cell. In the limit of a
large cell, this approximation becomes more and more accurate because
the majority of the occupied natural orbitals only extends over a
fraction of the simulation cell and therefore is not affected by the
boundary conditions. The justification for using this basis is that
one is generally interested in systems where the natural orbitals are
determined by the interactions rather than by boundary effects. However, in
systems where those are important, another basis that includes the cubic
symmetry is more appropriate. Suggestions have been made by
Shumway (1999).
The resulting matrices
![]() are then diagonalized,
which leads to natural orbitals as eigenvectors and eigenvalues
proportional to the occupation numbers. The relative occupation
numbers
are then diagonalized,
which leads to natural orbitals as eigenvectors and eigenvalues
proportional to the occupation numbers. The relative occupation
numbers ![]() are obtained by dividing the eigenvalues by the sum
of the traces of all
are obtained by dividing the eigenvalues by the sum
of the traces of all ![]() matrices. We found that the contributions
from different matrices decay rapidly with
matrices. We found that the contributions
from different matrices decay rapidly with ![]() and that keeping
matrices for
and that keeping
matrices for ![]() is more than sufficient.
is more than sufficient.
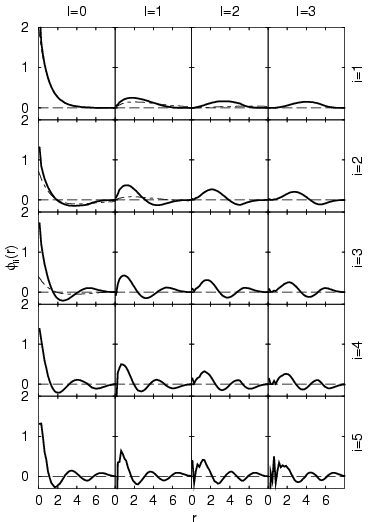
The resulting natural orbitals for a single hydrogen atom in a
periodically repeated box of size ![]() are shown in
Fig. 5.9. The displayed functions
are shown in
Fig. 5.9. The displayed functions
![]() are expected to approach the radial part
are expected to approach the radial part ![]() with
with ![]() of
the isolated hydrogen atom in the limit of a large box size. The
example reveals a ground state that is identical within statistical
and grid errors to the 1s ground state of the isolated hydrogen
atom. Studying the eigenvectors at any
of
the isolated hydrogen atom in the limit of a large box size. The
example reveals a ground state that is identical within statistical
and grid errors to the 1s ground state of the isolated hydrogen
atom. Studying the eigenvectors at any ![]() with increasing excitations
with increasing excitations
![]() , one finds that one additional node is introduced at each step
, one finds that one additional node is introduced at each step
![]() . The states with
. The states with ![]() are similar but not identical to those
of the isolated hydrogen atom because the finite size of the
simulation cell
are similar but not identical to those
of the isolated hydrogen atom because the finite size of the
simulation cell ![]() does have an effect, which leads to more
localized eigenstates. One also notices that the level of numerical noise
in the eigenvectors increases with
does have an effect, which leads to more
localized eigenstates. One also notices that the level of numerical noise
in the eigenvectors increases with ![]() . These effects seems to be the
strongest near the origin, which suggests that a different basis such
as hydrogen orbitals would lead to a lower noise level. Generally, one
finds that the noise in the eigenvectors (using the uniform spatial
grid) increases for lower temperatures because the occupation number
of higher energy states becomes very small. In this case, the
eigenvectors are approximately degenerate (
. These effects seems to be the
strongest near the origin, which suggests that a different basis such
as hydrogen orbitals would lead to a lower noise level. Generally, one
finds that the noise in the eigenvectors (using the uniform spatial
grid) increases for lower temperatures because the occupation number
of higher energy states becomes very small. In this case, the
eigenvectors are approximately degenerate (
![]() ) and
the noise causes that those states are mixed in the diagonalization
procedure. This explains why the noise level in the high eigenvectors
increase for lower temperature.
) and
the noise causes that those states are mixed in the diagonalization
procedure. This explains why the noise level in the high eigenvectors
increase for lower temperature.
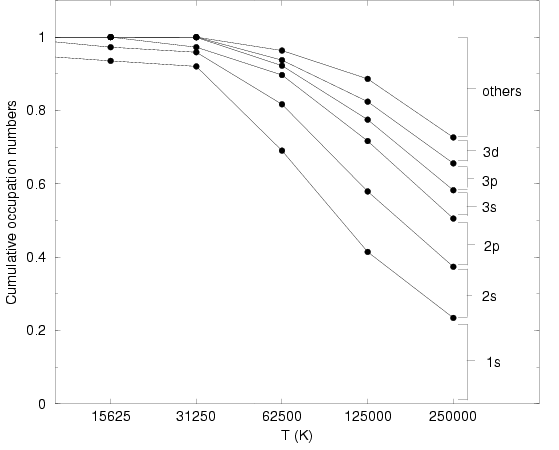 |
Studying the eigenvalues, one finds that there are a few large
positive ones while many others are small and some even negative. The
occupation numbers are shown in Fig. 5.10 as a function of
temperature. One finds that the occupation probability of the 1s
ground state increases with decreasing ![]() . Within the noise level of
about 4%, it goes to 1 in the limit of low
. Within the noise level of
about 4%, it goes to 1 in the limit of low ![]() . Furthermore, one finds
that the occupation of the 2s and 2p are almost the same despite the
fact that the 2 eigenvalues come from different matrices. The same
argument holds for the 3s, 3p, and 3d level. One can also calculate
the differences in energy from the ratio of the occupation
numbers. For
. Furthermore, one finds
that the occupation of the 2s and 2p are almost the same despite the
fact that the 2 eigenvalues come from different matrices. The same
argument holds for the 3s, 3p, and 3d level. One can also calculate
the differences in energy from the ratio of the occupation
numbers. For
![]() , one finds
, one finds ![]() rather
than
rather
than ![]() as expected for the isolated hydrogen atom. These
deviations increase if higher levels are studied because higher states
are more delocalized and therefore increasingly altered by the
boundary effects.
as expected for the isolated hydrogen atom. These
deviations increase if higher levels are studied because higher states
are more delocalized and therefore increasingly altered by the
boundary effects.