| Authors: | B. Militzer, F. Gonzalez-Cataldo, S. Zhang, K. P. Driver, F. Soubiran
| |
| |
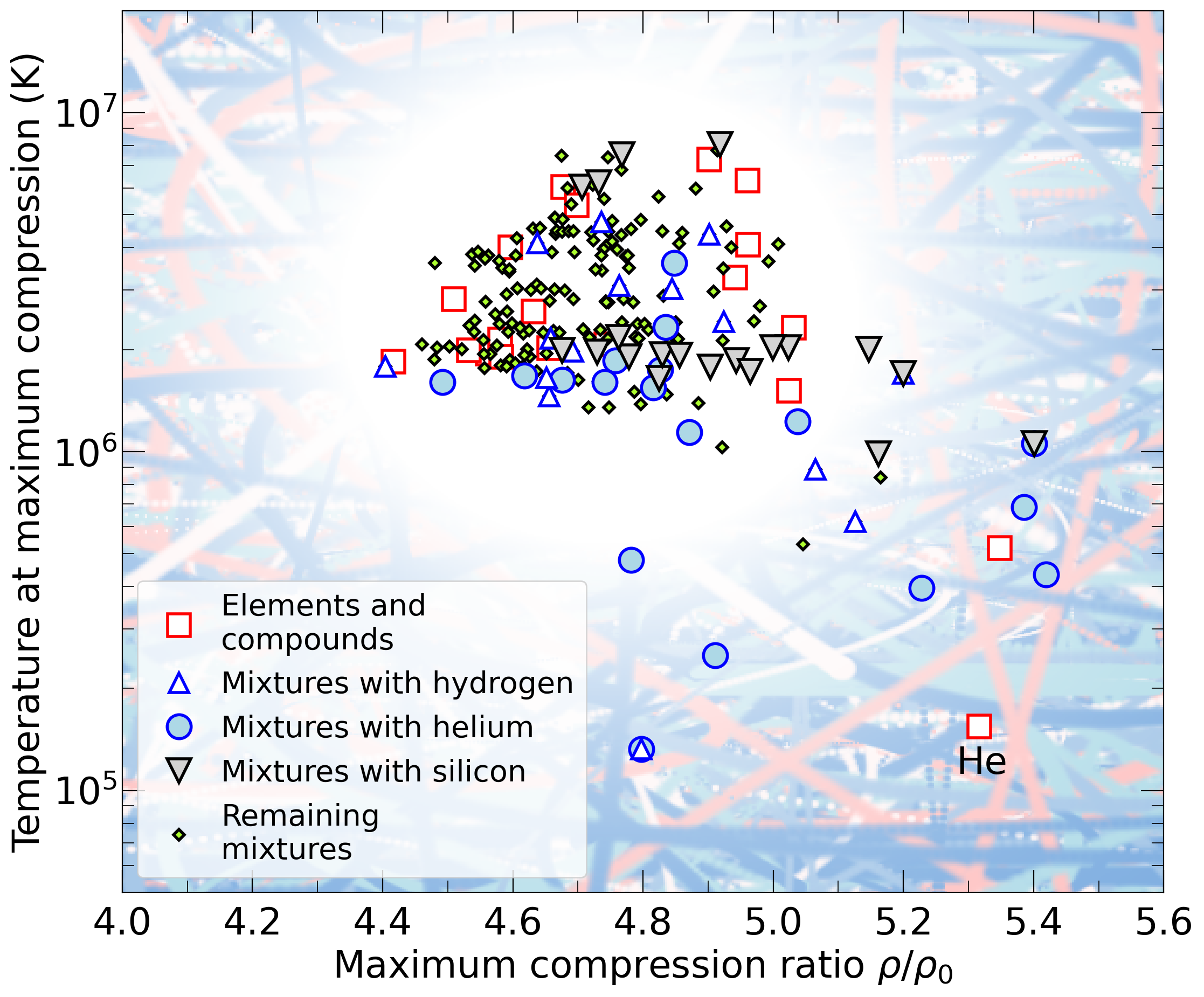
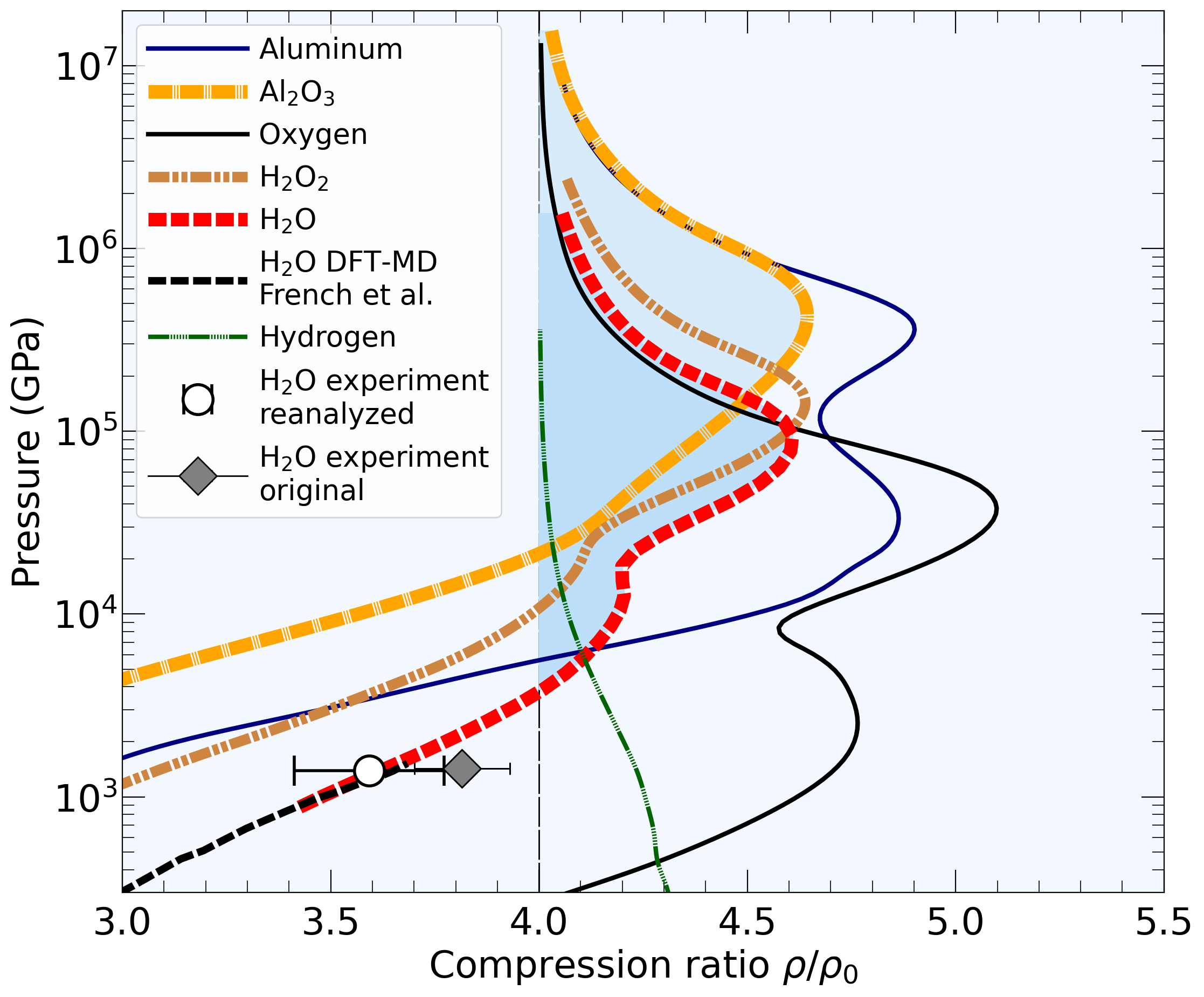
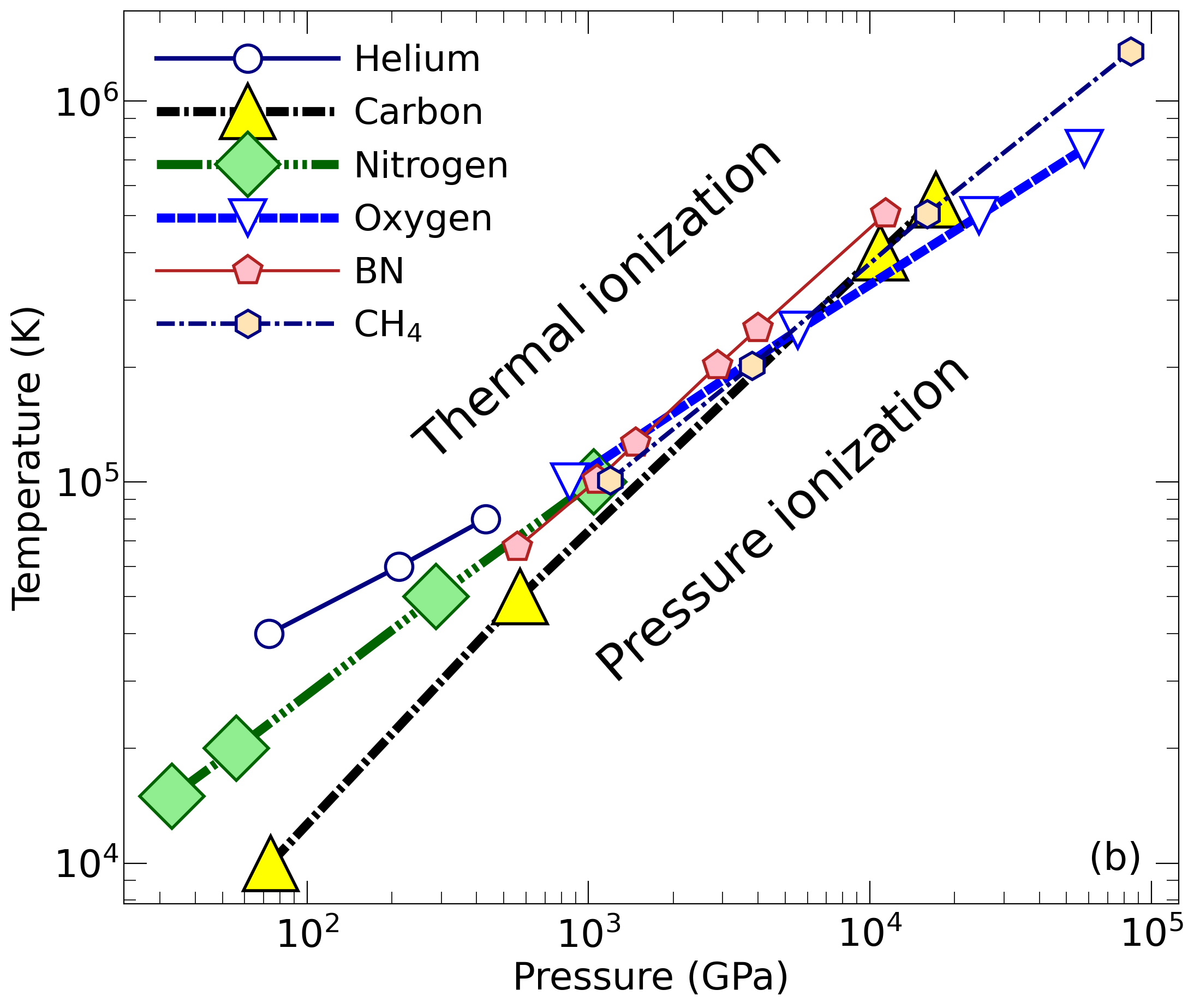
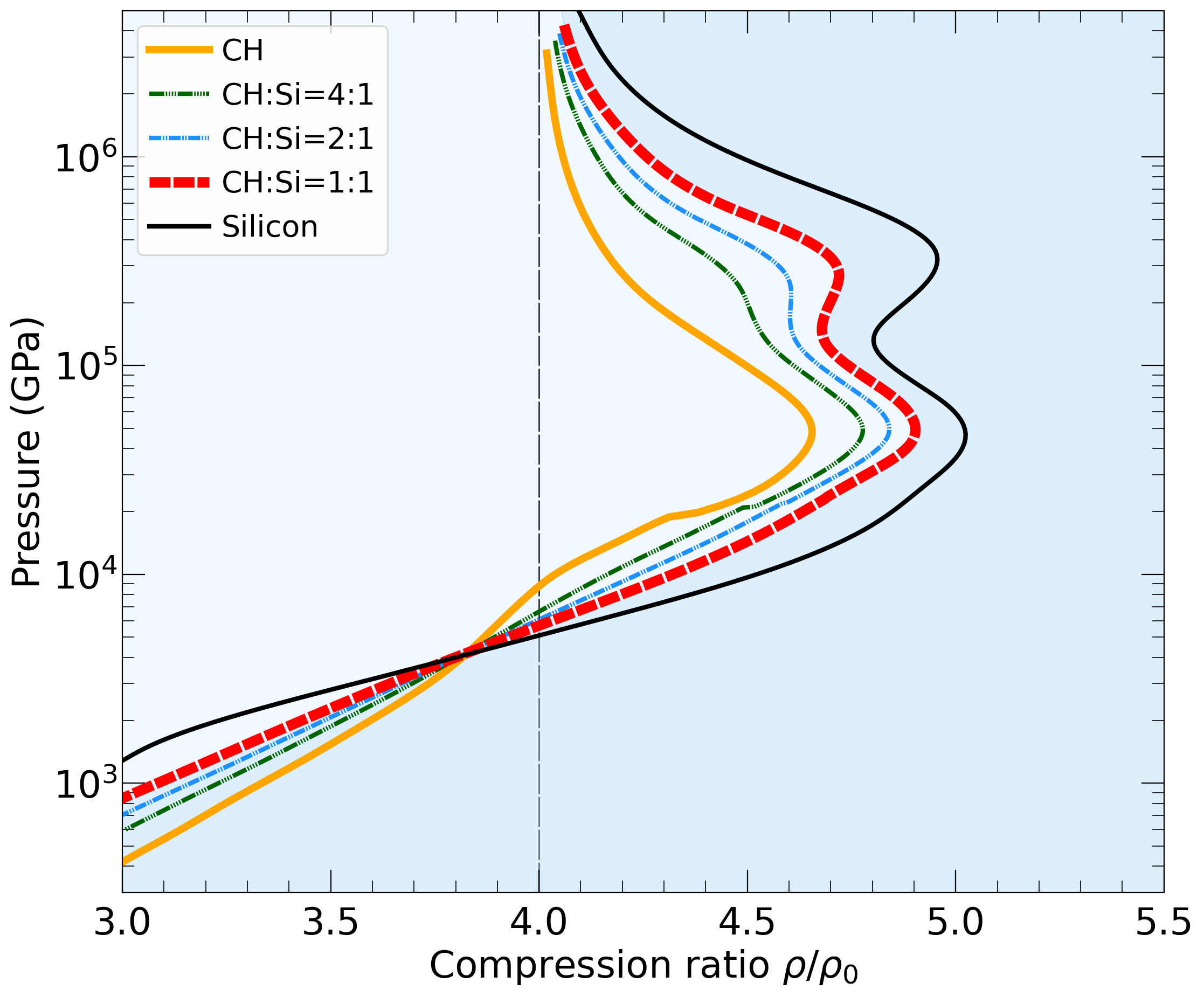
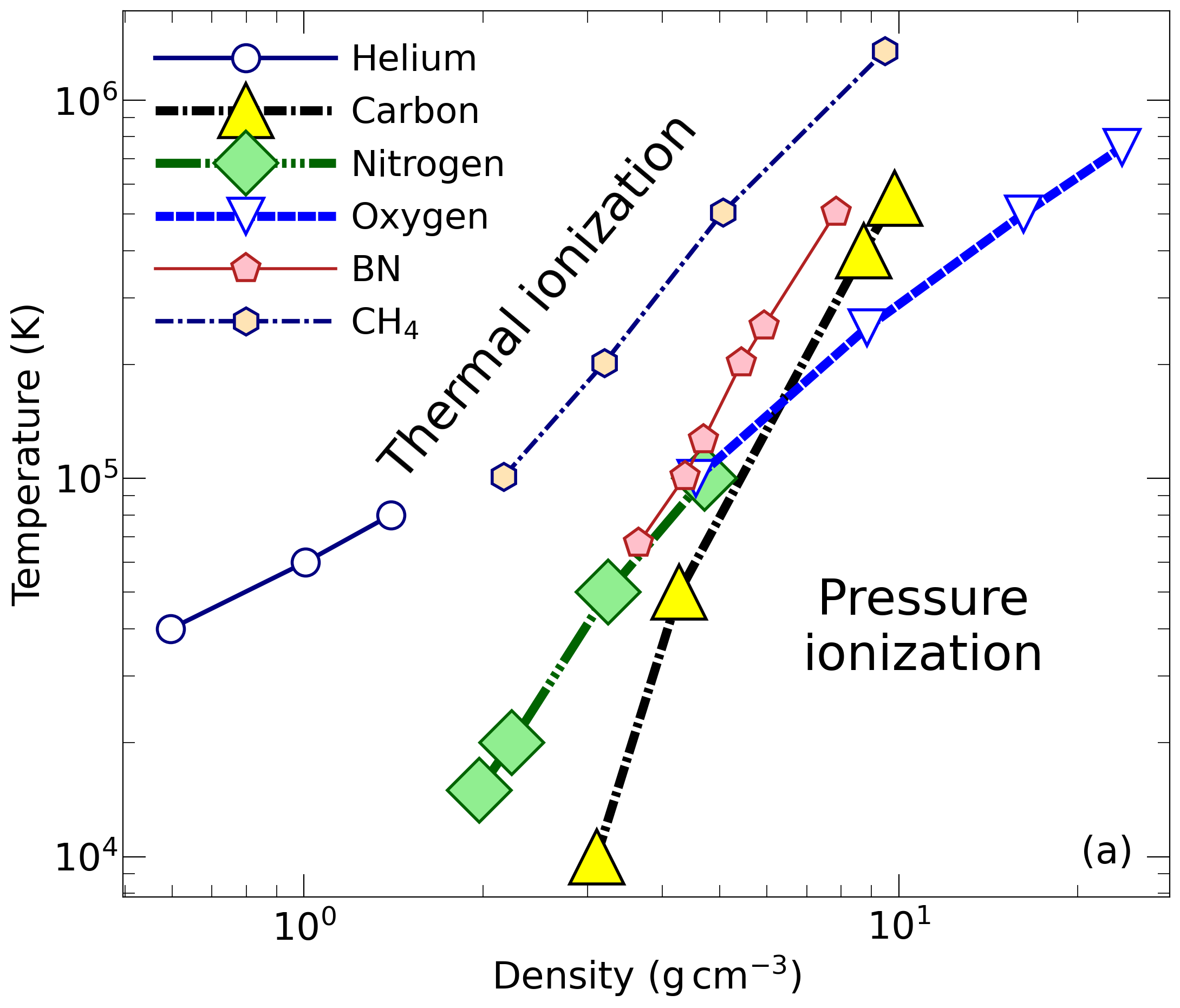
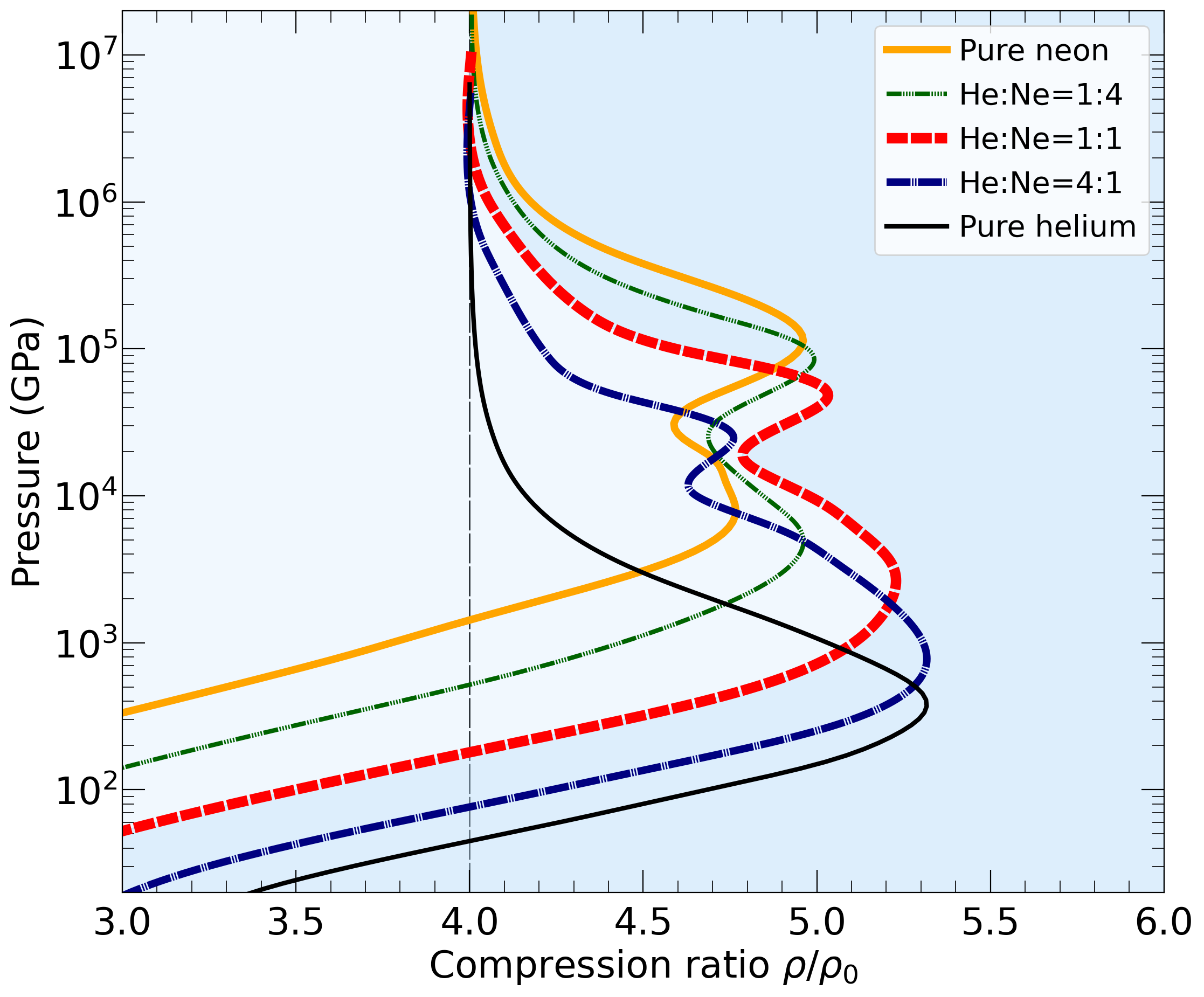
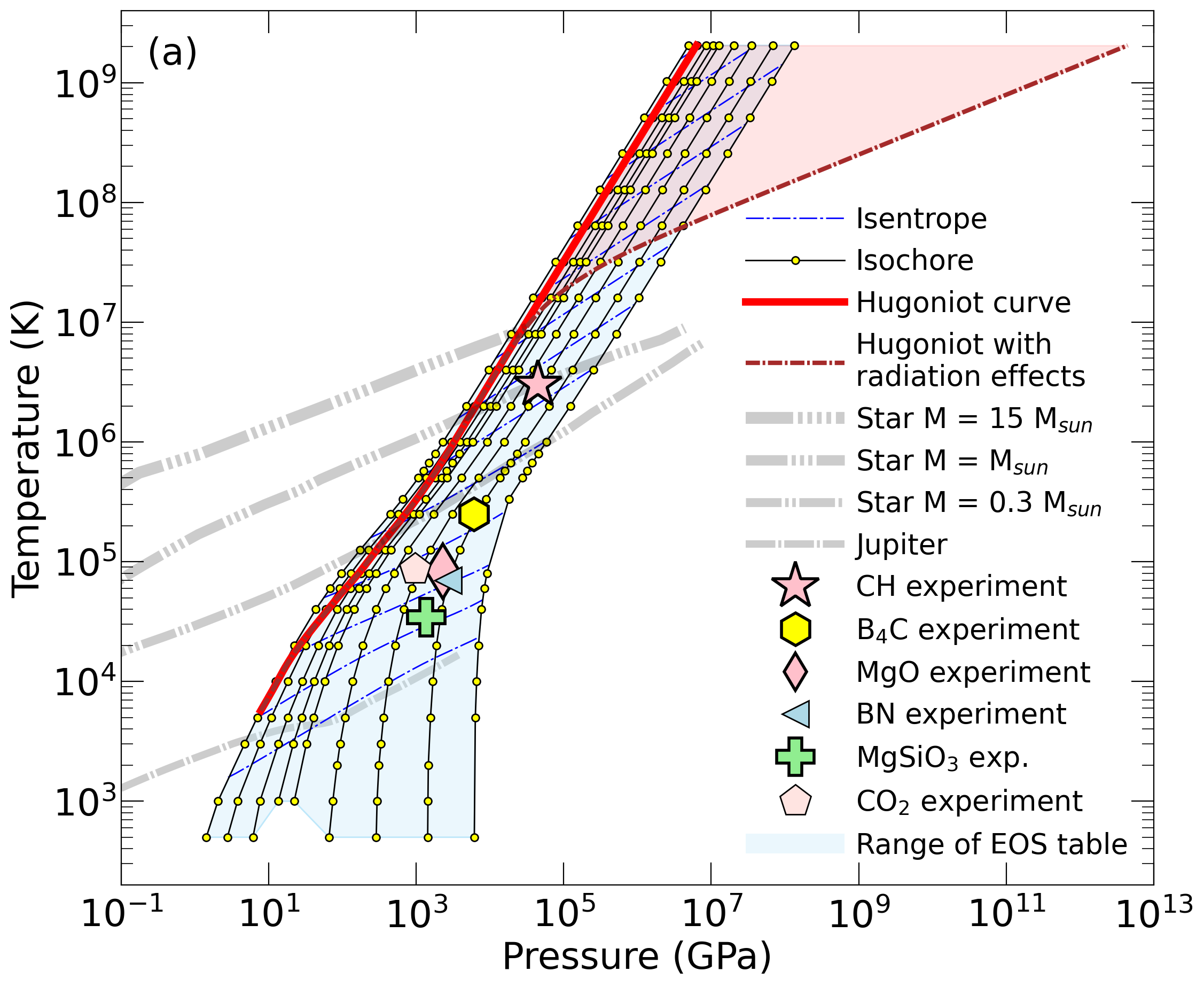
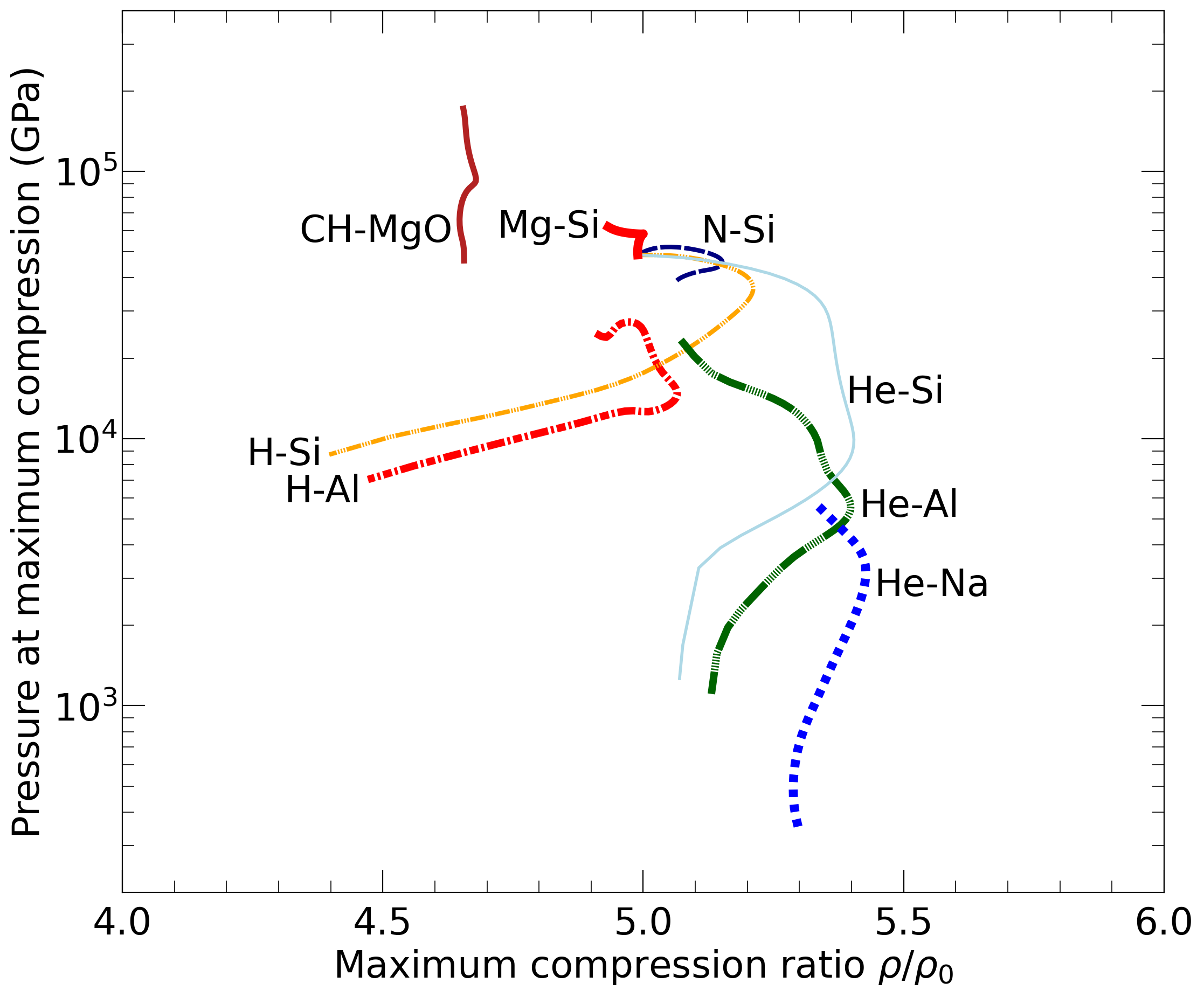
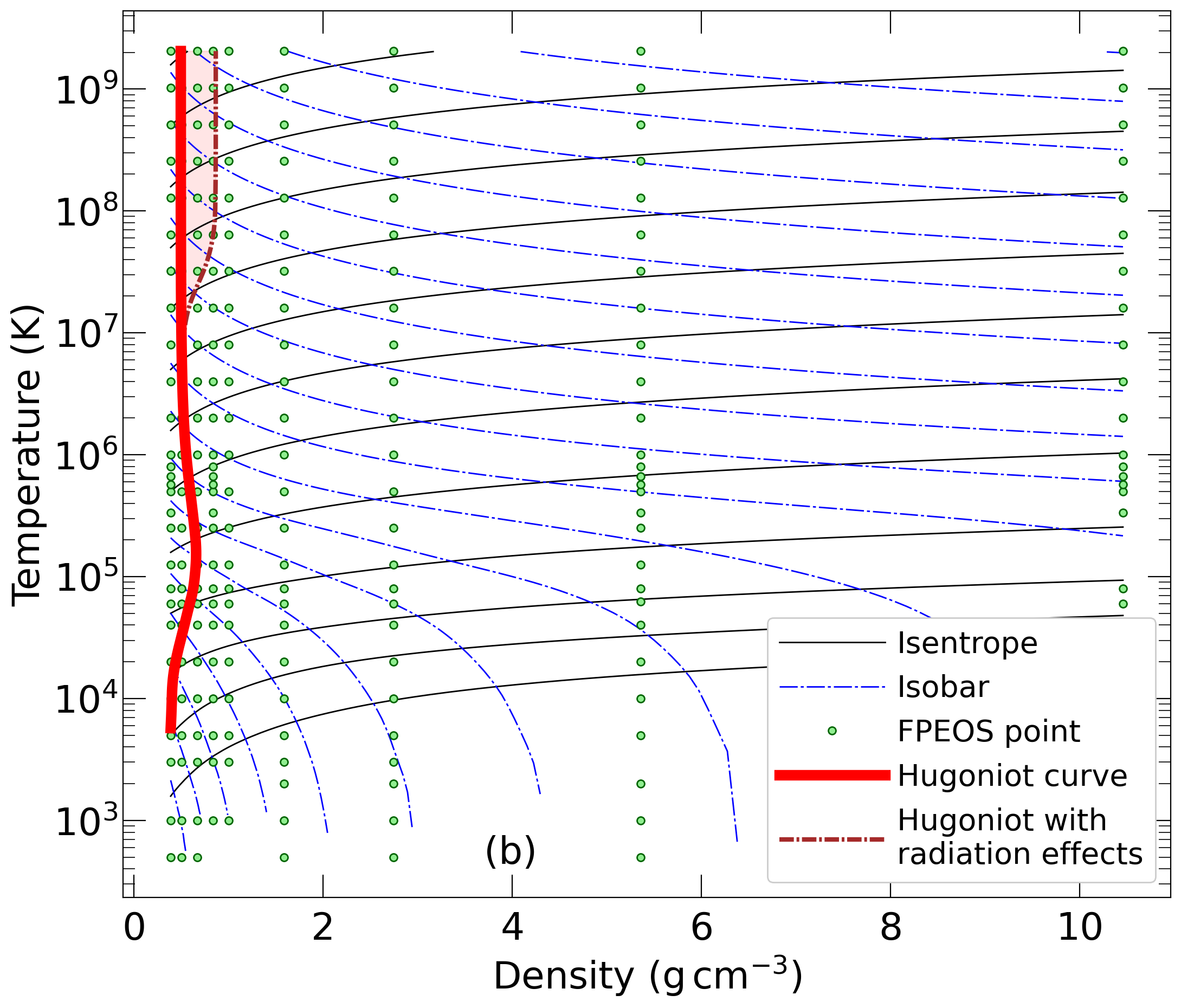
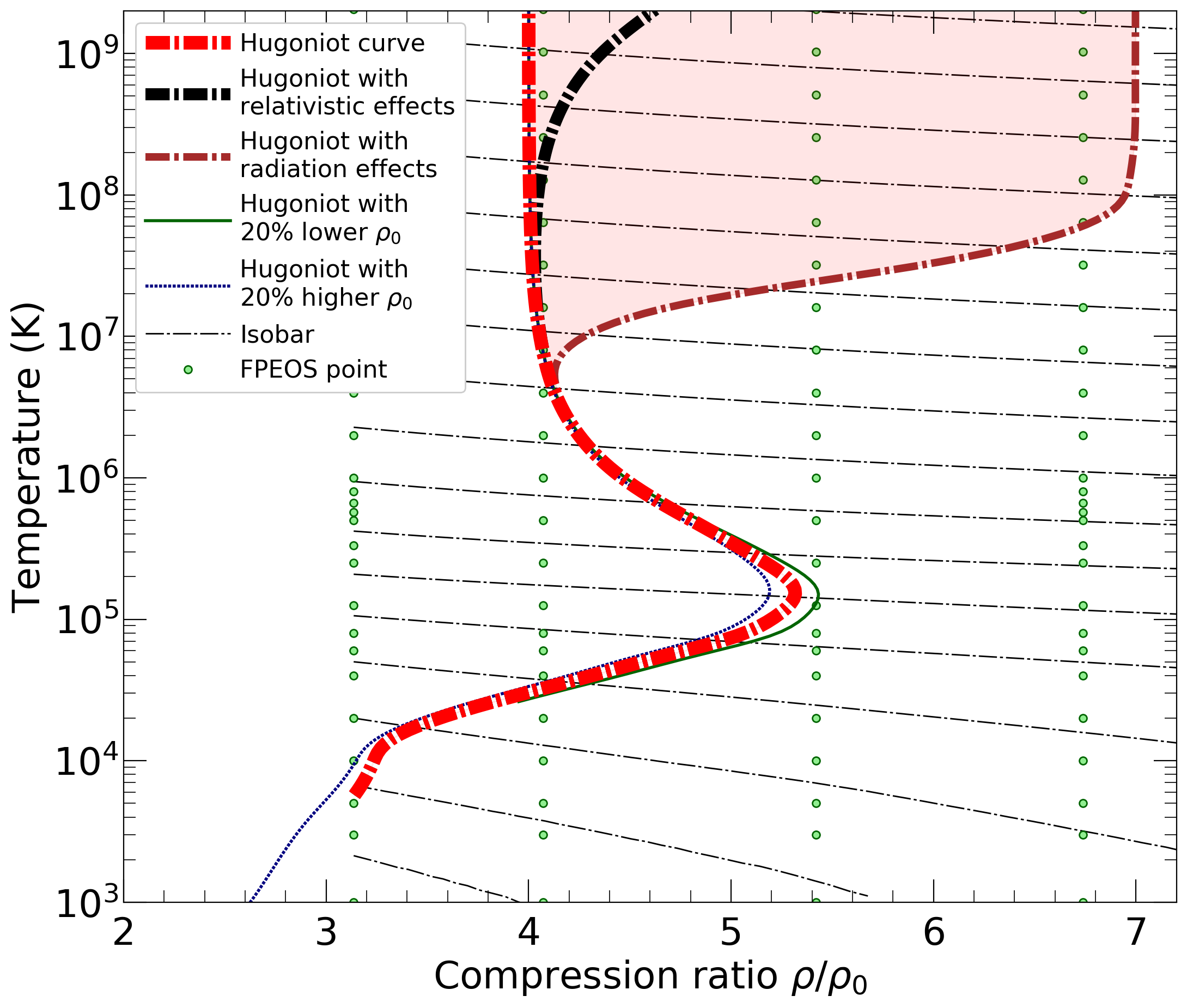
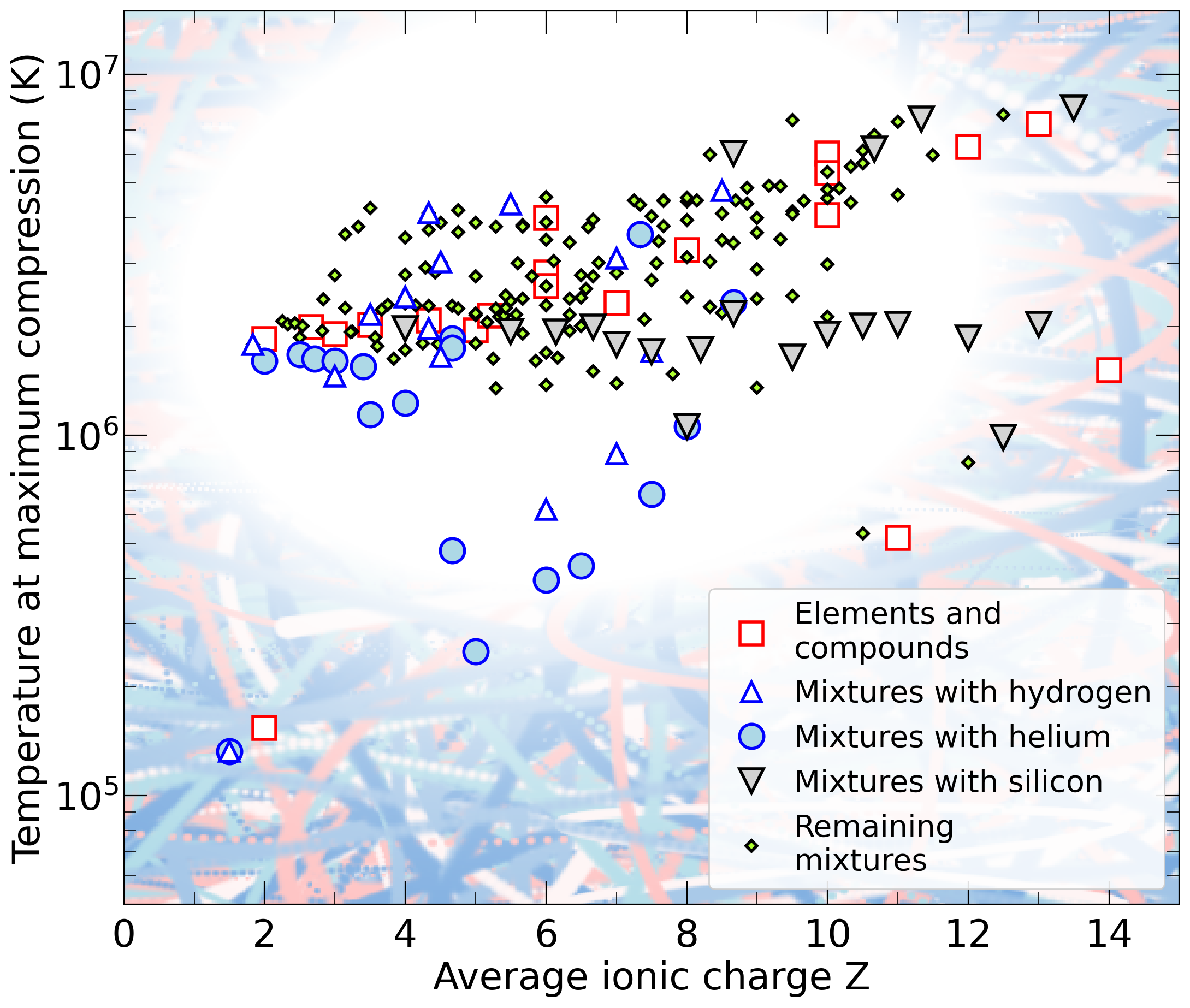
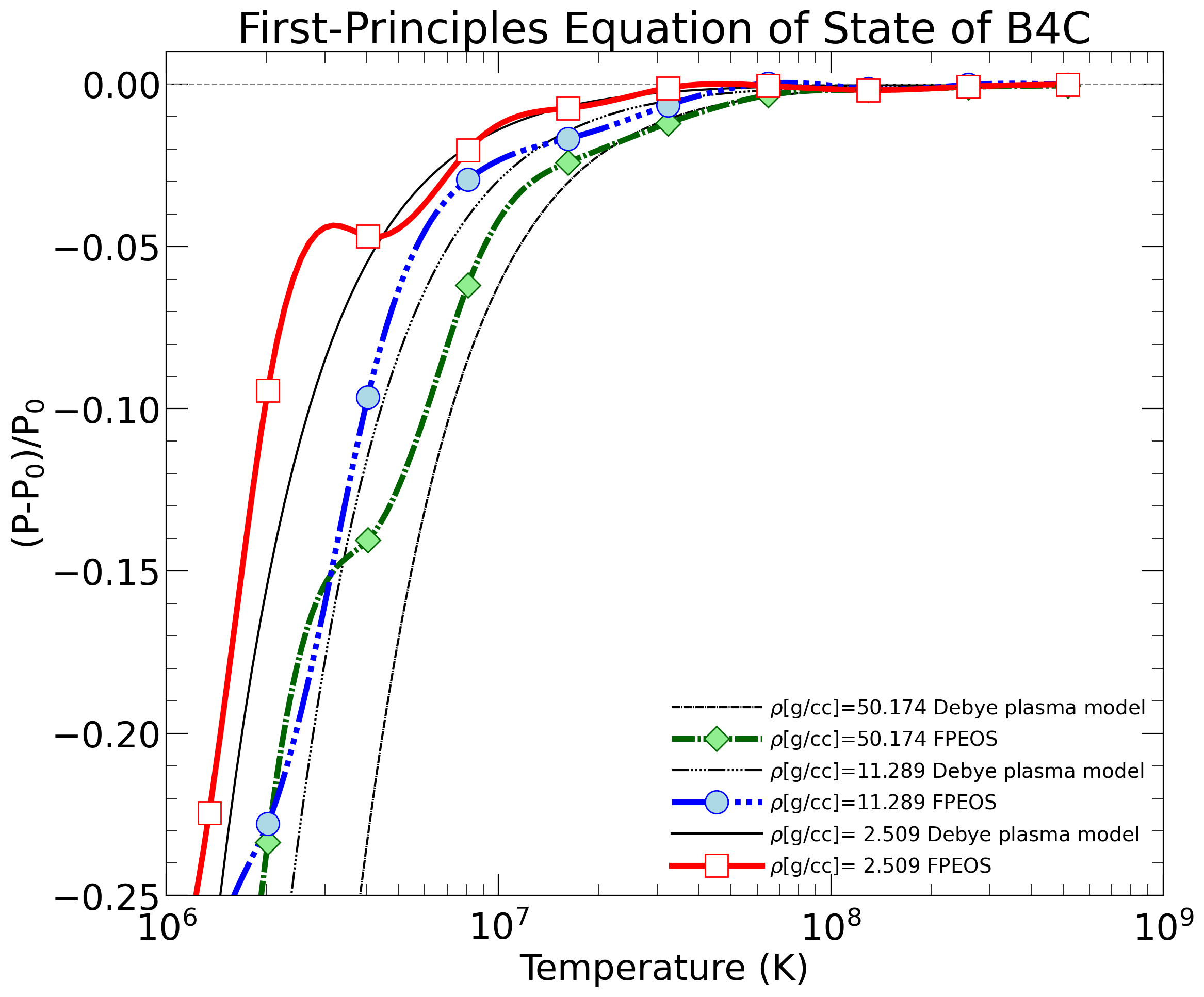
With the goal in mind of making WDM computations more reliable and efficient, we make available our EOS tables for 11 elements and 10 compounds as well as the C++ computer codes for their interpolation. Python code is provided to generate graphs of shock Hugoniot curve, isentropes, isobars, and isotherms for compounds and user-defined mixtures. We put together this first-principles equation of state (FPEOS) database for matter at extreme conditions by combining results from path integral Monte Carlo and density functional molecular dynamics simulations of the elements H, He, B, C, N, O, Ne, Na, Mg, Al and Si as well as the compounds LiF, B4C, BN, CH4, CH2, C2H3, CH, C2H, MgO, and MgSiO3. For all these materials, we provide the pressure and internal energy over a wide density-temperature range from ~0.5 to 50 g/cc and from ~104 to 109 K. This database encompasses the results from approximately 5000 different first-principles simulations. It allows one to compute isobars, adiabats, and shock Hugoniot curves in the regime of L and K shell ionization. Invoking the linear mixing approximation, one can study the properties of user-defined mixtures at high density and temperature.
Recommended citation: B. Militzer, F. Gonzalez-Cataldo, S. Zhang, K. P. Driver, F. Soubiran, "First-Principles Equation of State Database for Warm Dense Matter Computation", Physical Review E 103 (2021) 013203.
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
(1) Download the latest version
| Date |
File description | Download link |
| 05-22-24 | Figures are opened one by one. | fpeos_05-22-24.tar.gz |
| 05-21-24 | Added tutorial file: FPEOS_tutorial.pdf | fpeos_05-21-24.tar.gz |
| 05-20-24 | 1) For oxygen, set default rho0=1.33416818 2) Added openCommand=... and pythonCommand=... 3) Pylab -> PyPlot | fpeos_05-20-24.tar.gz |
| 05-17-24 | Minor updates in Python scripts | fpeos_05-17-24.tar.gz |
| 02-12-22 | This version supports ternary mixtures like C10-H16-O8 | fpeos_02-12-22.tar.gz |
| 01-07-21 | First public version of our FPEOS code. | fpeos_01-07-21.tar.gz |
(2) Installation
To install our FPEOS code, download and untar the latest version, change into the FPEOS directory and type make:
my_machine:~/Downloads> tar -xzf fpeos_01-07-21.tar.gz
my_machine:~/Downloads> cd FPEOS
my_machine:~/Downloads> make
which will call Gnu's C++ compiler g++ with the argument -O3. The output should be
g++ -O3 -c FPEOS_29.C
g++ -O3 -c EOSTableSimple.C
g++ -O3 -c Parser.C
g++ -O3 -c ReadInTable.C
g++ -O3 -c Standard.C
g++ -O3 -c Form.C
g++ -O3 -c Physics.C
g++ -O3 -c Spline.C
g++ -O3 -c FermiGas.C
g++ -O3 -c RelativisticFermiGas.C
g++ -O3 -c Debye.C
g++ -O3 -c GKIntegration.C
g++ -O3 -c RungeKutta.C
g++ FPEOS_29.o EOSTableSimple.o Parser.o ReadInTable.o Standard.o Form.o Physics.o Spline.o FermiGas.o RelativisticFermiGas.o Debye.o GKIntegration.o RungeKutta.o -lm -o fpeos
In case this fails, try editing the Makefile or switch to a different compiler.
We do not provide executables for any operating systems here but our source code has been compiled successfully with these three compilers:
Linux g++ version 7.5.0 (Ubuntu 7.5.0-3ubuntu1~18.04)
Linux g++ version 4.8.4 (Ubuntu 4.8.4-2ubuntu1~14.04.4)
macOS clang version 13.0.0 (clang-1300.0.29.30)
macOS clang version 12.0.0 (clang-1200.0.32.2)
To run our code, you need a C++ compiler. Python needs to be installed to generate the graphs (but C++ code will work without Python). Linux machines typically have the g++ compiler and Python already installed.
On Macs, we recommend installing XCode with the command line tools as well as installing Python with Anaconda (but there could be more disk space conserving options to install a C++ compiler and Python).
On Windows, install Python as well as Cygwin with g++ version 10.2.1. Compile and run the resulting fpeos.exe. Open the PDF files with cygstart.
(3) Files included
The EOS tables are stored in files
In the every file, you find a header followed by blocks of data, one for every density. Our carbon EOS file begins with
f= C N= 1 rho[g/cc]= 0.100006 V[A^3]= 199.43071700 T[K]= 5000 P[GPa]= 0.053 0.007 E[Ha]= -37.66843800 0.00054900
f= C N= 1 rho[g/cc]= 0.100006 V[A^3]= 199.43071700 T[K]= 10000 P[GPa]= 0.182 0.022 E[Ha]= -37.56017000 0.01215800
f= C N= 1 rho[g/cc]= 0.100006 V[A^3]= 199.43071700 T[K]= 50000 P[GPa]= 5.670 0.013 E[Ha]= -36.69107600 0.00495700
| 2) | The chemical formula, such as C, MgSiO3, C10H40, etc. This entry is not used to normalized V and E. |
| 4) | Number of atoms, N, that are used to normalized the extensive quantities V and E in later columns. |
| 6) | Density in gramm/cm3 using standard masses for all atoms. |
| 8) | Volume of N atoms in units of Angstroem3 (1 Angstroem = 10-10 meters = 1/0.529177208607 aBohr). This is directly taken from the simulations and does not depend on the assumed atomic masses. |
| 10) | Temperature in units of Kelvin (K). |
| 12) | Pressure derived from PIMC or DFT-MD simulations in units of 109 Pascals. |
| 13) | 1-sigma error bar of pressure that was derived when the ensemble average was computed. |
| 15) | Internal energy per N atoms in units of Hartrees (1 Ha = 4.359743802665602 * 10-18 J = 27.21138342791109 eV = 315774.6608728946 K * kb). The zero of energy is that of a fully ionized system so that an hydrogen atom has the energy of -0.5 Ha = -13.6 eV. The means all VASP energies need to be shifted but those from Cupid, Abinit, Quantum Espresso, and most quantum chemistry codes do not. |
| 16) | 1-sigma error bar of the internal energy that was derived when the ensemble average was computed. |
The C++ files are
The C++ code calls the following Python scripts to write graphics files in .png and .pdf formats.
These files should work with Python 2.7 and 3.7 as long as the following import commands work:
from matplotlib.pylab import *
import numpy
import sys
import os
Finally there is a "readme.txt" file.
(4) Execution
To obtain the latest instructions type
fpeos
which will print the elements and compounds in the database:
| 1: Hydrogen | 8: Sodium | 15: CH4 |
| 2: Helium | 9: Magnesium | 16: CH2 |
| 3: Boron | 10: Aluminum | 17: CH1.5 |
| 4: Carbon | 11: Silicon | 18: CH |
| 5: Nitrogen | 12: LiF | 19: CH0.5 |
| 6: Oxygen | 13: B4C | 20: MgO |
| 7: Neon | 14: BN | 21: MgSiO3 |
You can run our fpeos code in the following ways:
| For helium EOS: | fpeos EOS=2 |
| For B4C EOS: | fpeos EOS=13 |
| For MgSiO3 EOS: | fpeos EOS=21 |
| For H+He mixture: | fpeos binaryMixture EOS1=1 0.92 EOS2=2 0.08 |
| For He+Ne mixture: | fpeos binaryMixture EOS1=2 1.0 EOS2=7 1.0 |
| For MgO+MgSiO3 mixture: | fpeos binaryMixture EOS1=20 1.0 EOS2=21 1.0 |
| To study carbon monoxide CO ≈ C+O mixture: | fpeos binaryMixture EOS1=4 1.0 EOS2=6 1.0 rho0=1.0426 E0=-112.9115 |
| To study carbon dioxide CO2 ≈ C+2O mixture: | fpeos binaryMixture EOS1=4 1.0 EOS2=6 2.0 rho0=1.6381 E0=-188.1576 |
| To study water H2O ≈ 2H+O mixture: | fpeos binaryMixture EOS1=1 2.0 EOS2=6 1.0 rho0=1.0 E0=-76.392172 |
| To study hydrogen peroxide H2O2 ≈ 2H+2O mixture: | fpeos binaryMixture EOS1=1 2.0 EOS2=6 2.0 rho0=1.71330 E0=-151.4893 |
| To study alumina Al2O3 ≈ 2Al+3O mixture: | fpeos binaryMixture EOS1=10 2.0 EOS2=6 3.0 rho0=3.9929 E0=-708.8067 |
| To study acetic acid C2O2H4 ≈ 2CH2+2O mixture: | fpeos binaryMixture EOS1=6 2.0 EOS2=16 2.0 rho0=1.049 E0=-229.0 |
| To study glyoxal C2O2H2 ≈ 2CH +2O mixture: | fpeos binaryMixture EOS1=6 2.0 EOS2=18 2.0 rho0=1.27 E0=-227.8 |
rho0[g/cc] and E0[Ha/FU] overwrite the default values for initial conditions of Hugoniot curves.
The extra numbers are number fractions that define the formula unit (FU), which is used to normalize extensive quantities like volume and energy.
| To normalize per C2H6 unit, write: | fpeos binaryMixture EOS1=15 1.0 EOS2=16 1.0 |
| To normalize per CH3 unit, write: | fpeos binaryMixture EOS1=15 0.5 EOS2=16 0.5 |
| To normalize per atom, write: | fpeos binaryMixture EOS1=15 0.125 EOS2=16 0.125 |
The FPEOS code will write a number of .txt files, then call Python to make plots, and finally try to open the .pdf files. To identify all new output files, type "ls -ltr"
Add the options "doNotCallPython" or "doNotOpenPDFFiles" if you prefer. The arguments "processAllEOSs", "processAllMixturesOfElements", "processAllMixtures", "hugoniotCompressionMaximum", and "noLogInterpolation" are available also.
(5) Resulting files
In standard Linux fashion, type "ls -ltr" to figure out, which files have been written.
For "fpeos EOS=13", the result should be
{kind=link}
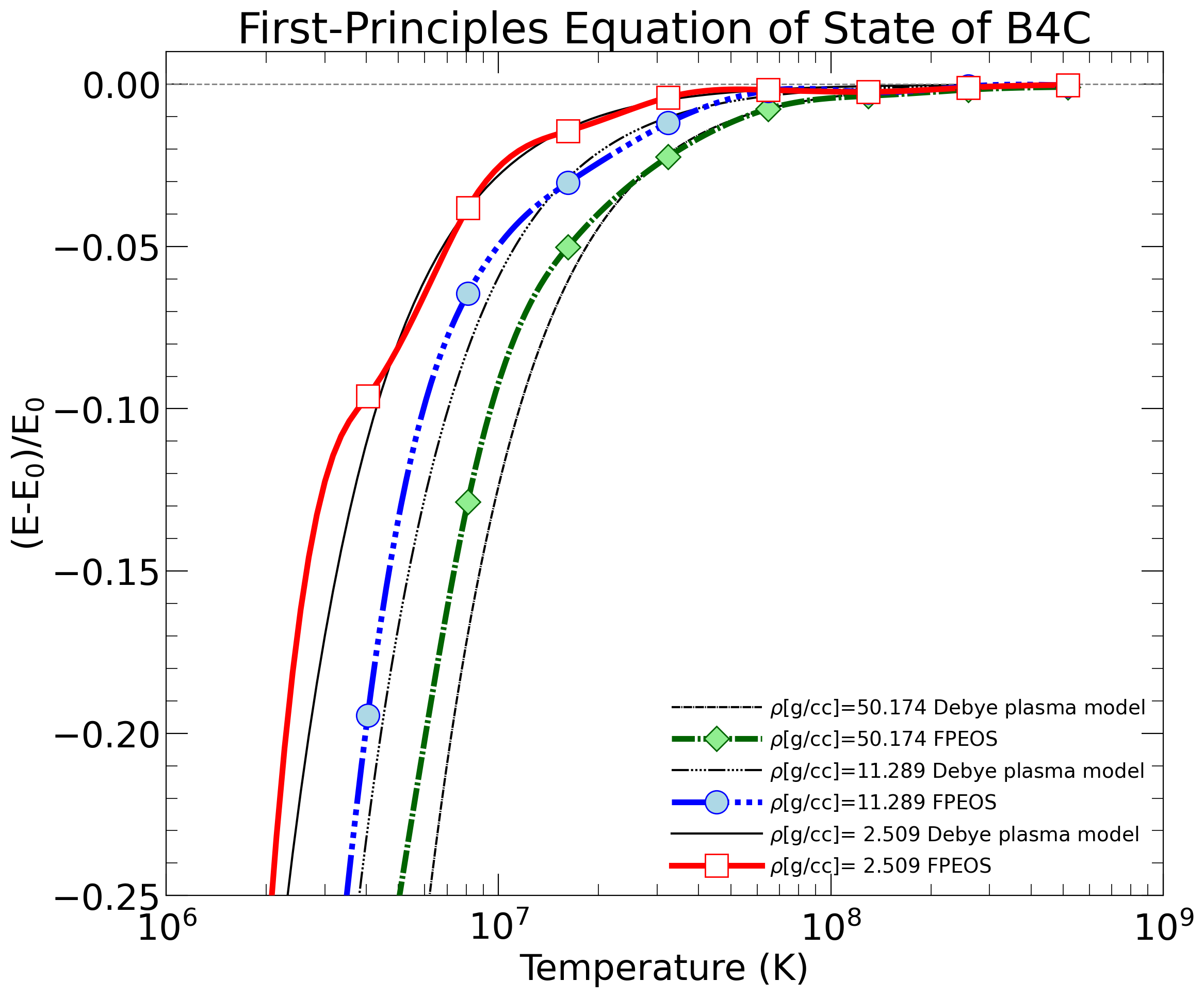{kind=link}
{kind=link}
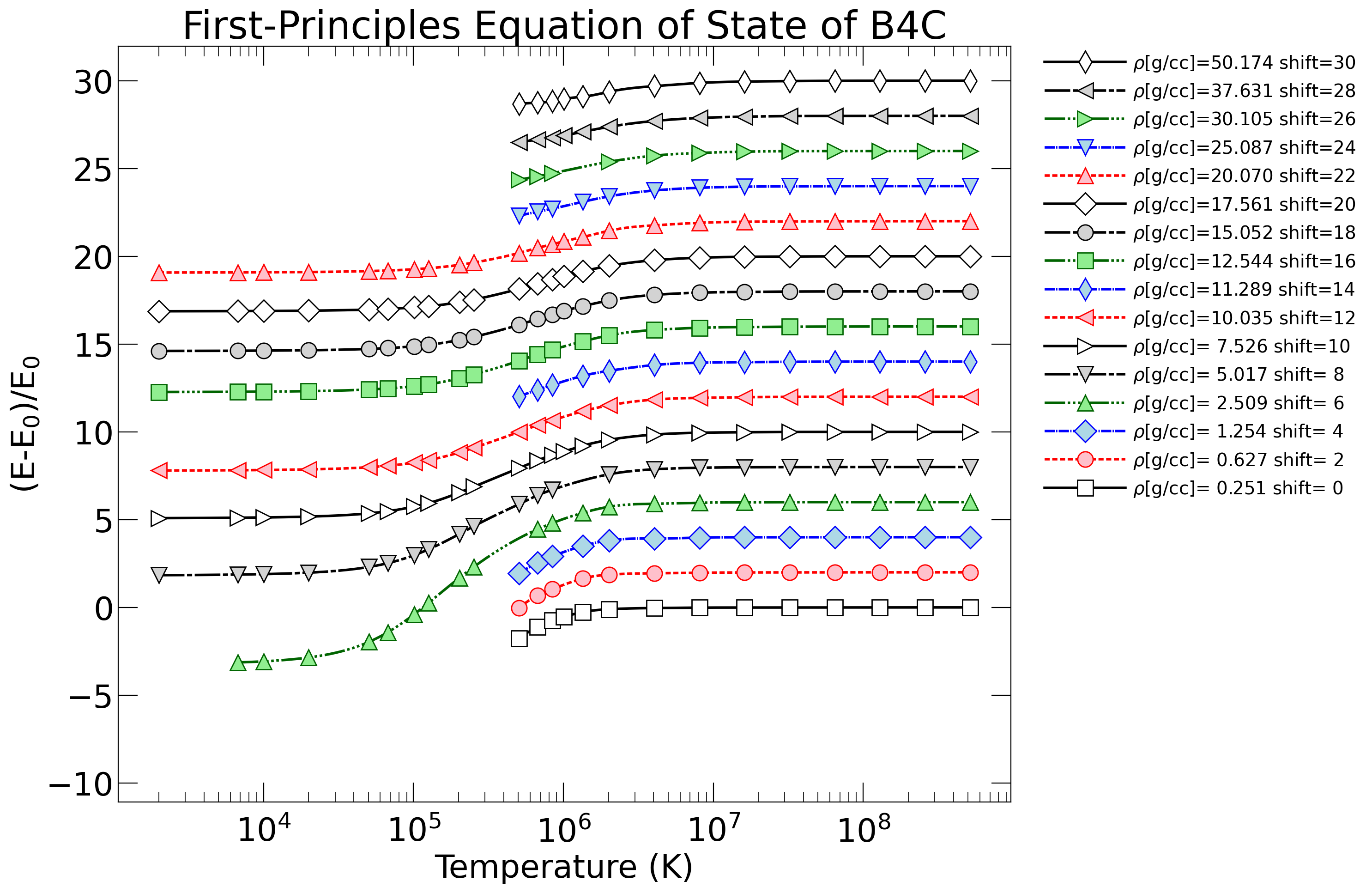{kind=link}
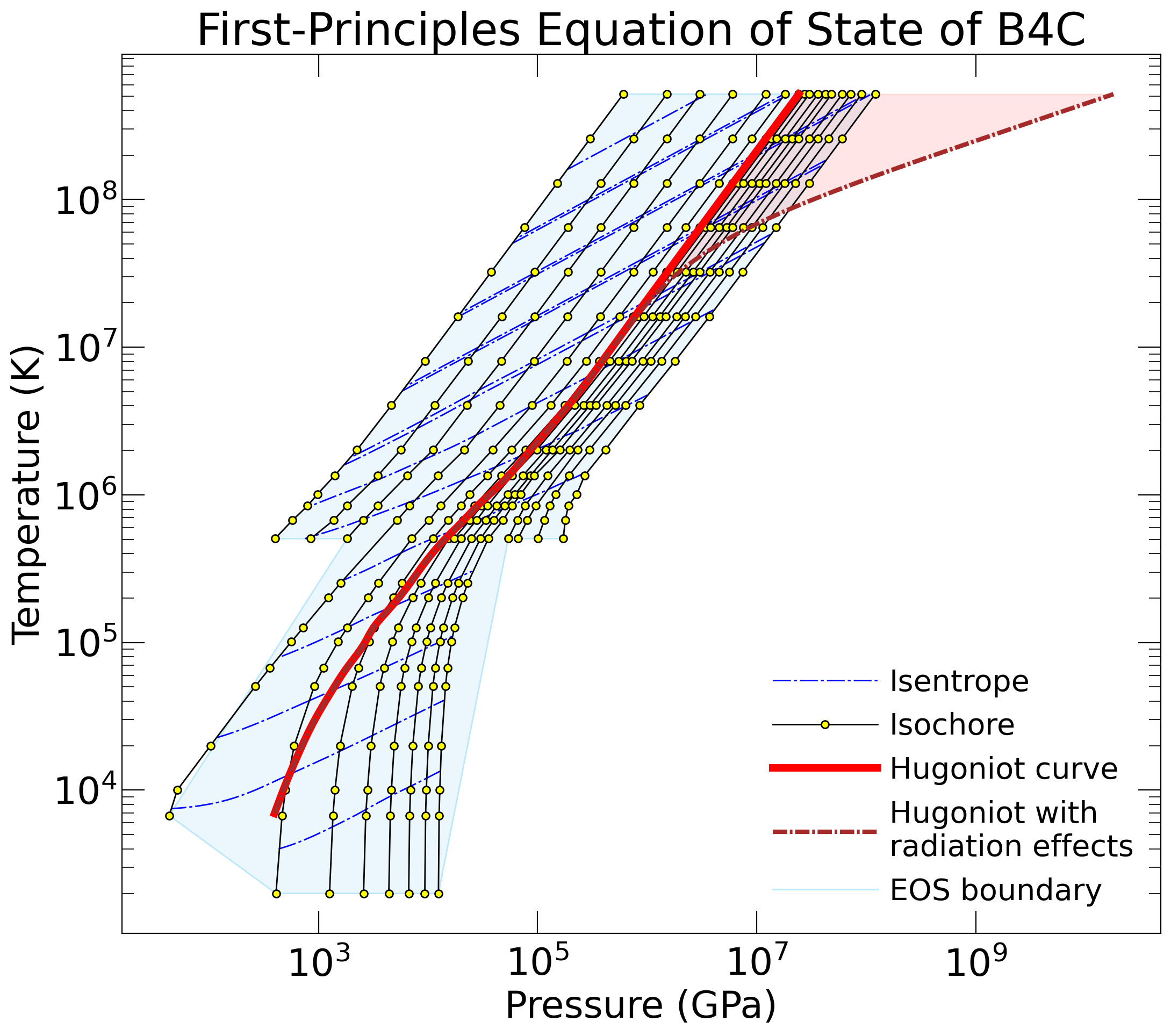{kind=link}
The .txt files were all written by the fpeos executable. The .png and .pdf files were produced by the Python scripts that fpeos called.
(6) A few concluding remarks
| 1) | As initial conditions of the Hugoniot curve of a mixture, the code will assume a non-bonding, heterogenous mixture of compounds A and B. By default, both compounds are assumed in the initial state that we studied in the original publication. For oxygen, this was the crystalline Γ phase at a density of ρ0=0.667 g/cc. To study H2O, one needs to overwrite the initial density and internal energy, E0, to include the effects of chemical bonding. We cannot provide the E0 value for all possible starting states. Any all-electron DFT code can be used to derive a reasonable E0 but admittedly this requires some additional skills. However, such calculations do not need to be perfect and are simpler than computing the P(ρ,T) and E(ρ,t) data points in our database. |
| 2) | E0 needs to be given in Hartrees per formula unit (FU). For compounds, the FU is defined in the tables (e.g. For oxygen this is an atom, not a molecule. For ethylenyl, it is CH1.5 instead of C2H3.) For mixtures, FU is defined by two parameters in the command line. When they are changed, E0 must be changed accordingly. |
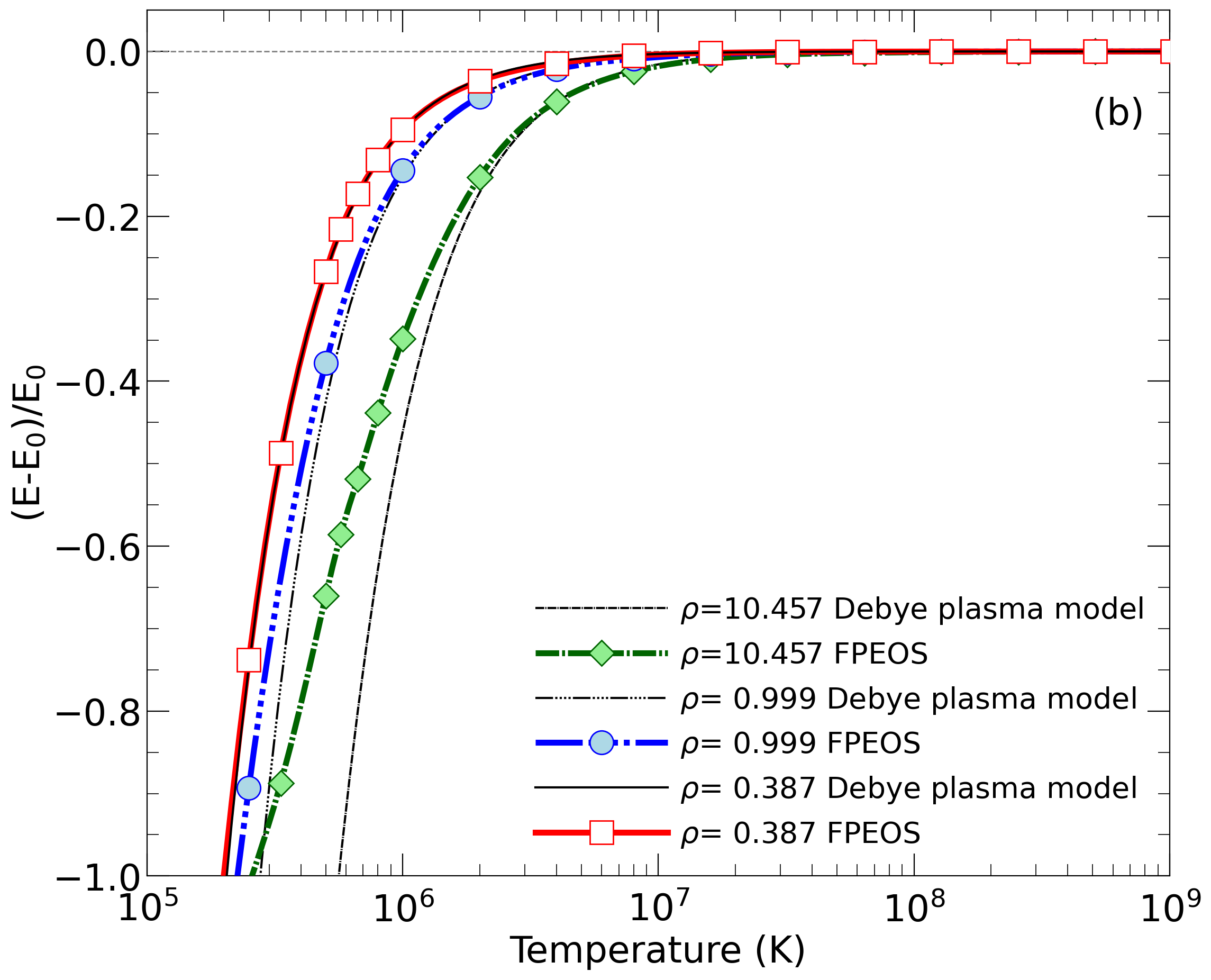
| 3) | Within our C++ code, we calculate properties an ideal Fermi gas and a Debye plasma. Anyone is welcome to incorating these routines elsewhere. |
| 4) | I wrote these C++ routines over several years to perform day-to-day calculations. They were kept simple and big libraries have been avoided. |
| 5) | Any regular Python user will find our plotting routines straightforward. Anyone else may find the quality an improvement over earlier plotting software. |
If you have feedback, comments, or a specific scientific problem you would like to solve please send email to militzer[at]berkeley[dot]edu.
Best wishes and happy FPEOS computation!
Burkhard Militzer
May 20, 2024.